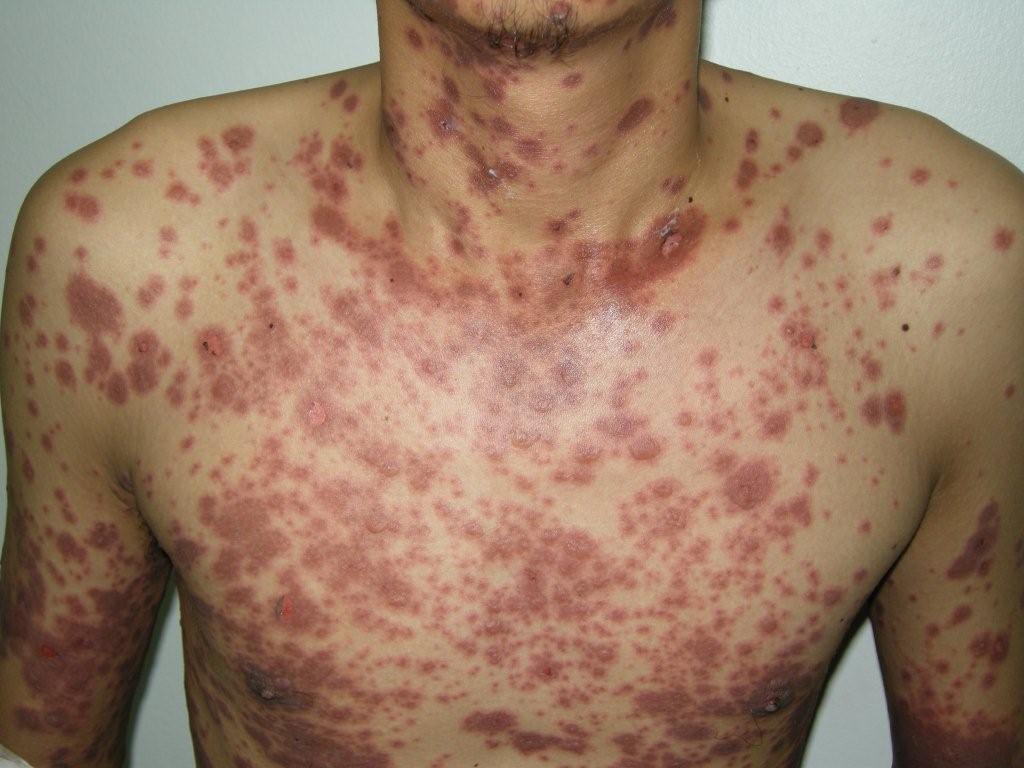
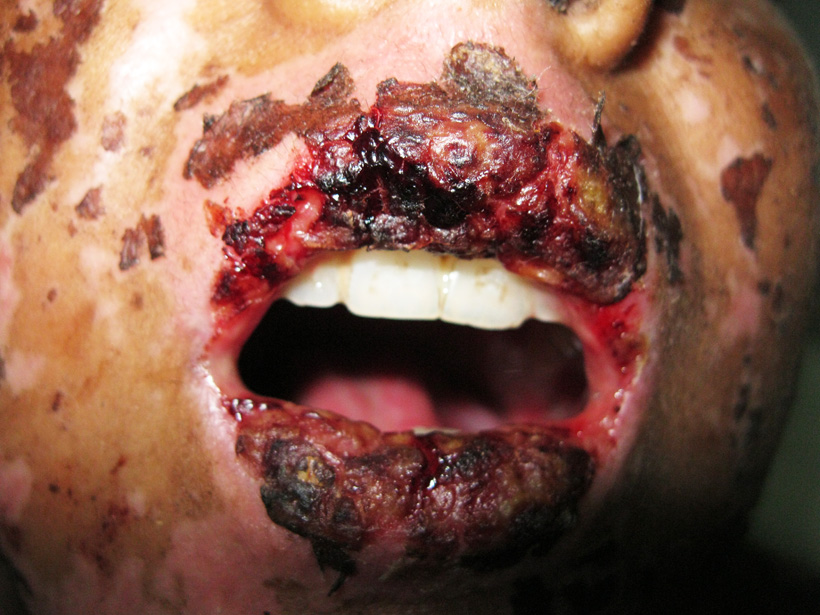
Stevens-Johnson syndrome is a rare, serious disorder of your skin and mucous membranes. It’s usually a reaction to a medication or an infection. Often, Stevens-Johnson syndrome begins with flu-like symptoms, followed by a painful red or purplish rash that spreads and blisters. Then the top layer of the affected skin dies and sheds.
Stevens-Johnson syndrome is a medical emergency that usually requires hospitalization. Treatment focuses on eliminating the underlying cause, controlling symptoms and minimizing complications.
Recovery after Stevens-Johnson syndrome can take weeks to months, depending on the severity of your condition. If it was caused by a medication, you’ll need to permanently avoid that drug and others closely related to it.
Stevens-Johnson syndrome is an immune-complex–mediated hypersensitivity complex that typically involves the skin and the mucous membranes. Although several classification schemes have been reported, the simplest classification breaks the disease down as follows:
- Stevens-Johnson syndrome: A minor form of toxic epidermal necrolysis, with less than 10% body surface area (BSA) detachment
- Overlapping Stevens-Johnson syndrome/toxic epidermal necrolysis: Detachment of 10-30% of the BSA
- Toxic epidermal necrolysis: Detachment of more than 30% of the BSA
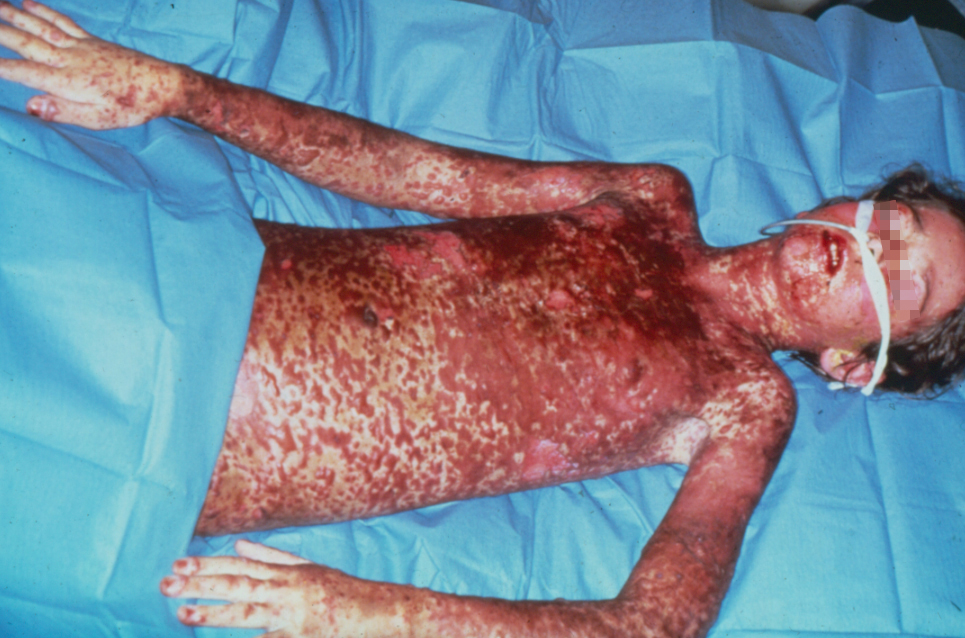
Signs and symptoms
Typical prodromal symptoms of Stevens-Johnson syndrome are as follows:
- Cough productive of a thick, purulent sputum
- Headache
- Malaise
- Arthralgia
Patients may complain of a burning rash that begins symmetrically on the face and the upper part of the torso. The cutaneous lesions are characterized as follows:
- The rash can begin as macules that develop into papules, vesicles, bullae, urticarial plaques, or confluent erythema
- The typical lesion has the appearance of a target; this is considered pathognomonic
- In contrast to the typical lesions of erythema multiforme, these lesions have only 2 zones of color
- The lesion’s core may be vesicular, purpuric, or necrotic; that zone is surrounded by macular erythema
- Lesions may become bullous and later rupture, leaving denuded skin; the skin becomes susceptible to secondary infection
- Urticarial lesions typically are not pruritic
- Infection may be responsible for the scarring associated with morbidity
- Although lesions may occur anywhere, the palms, soles, dorsum of the hands, and extensor surfaces are most commonly affected
- The rash may be confined to any one area of the body, most often the trunk
Signs of mucosal involvement can include the following:
- Erythema
- Edema
- Sloughing
- Blistering
- Ulceration
- Necrosis
The following ocular signs may be noted on slit-lamp examination:
- Eyelids: Trichiasis, distichiasis, meibomian gland dysfunction, blepharitis
- Conjunctiva: Papillae, follicles, keratinization, subepithelial fibrosis, conjunctival shrinkage, foreshortening of fornices, symblepharon, ankyloblepharon
- Cornea: Superficial punctate keratitis, epithelial defect, stromal ulcer, neovascularization, keratinization, limbitis, conjunctivalization, stromal opacity, perforation
Diagnosis
Minimal dermal inflammatory cell infiltrate and full-thickness necrosis of the epidermis are typical histopathologic findings in patients with Stevens-Johnson syndrome. Histopathologic examination of the skin can also reveal the following:
- Changes in the epidermal-dermal junction ranging from vacuolar alteration to subepidermal blisters
- Dermal infiltrate: Superficial and mostly perivascular
- Apoptosis of keratinocytes
- CD4+ T lymphocytes predominating in the dermis; CD8+ T lymphocytes predominating in the epidermis; the dermoepidermal junction and epidermis is infiltrated mostly by CD8+ T lymphocytes
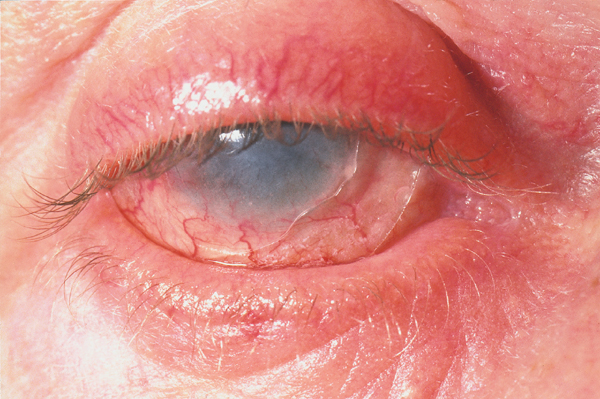
Ocular examination can demonstrate the following:
- Conjunctival biopsies from patients with active ocular disease show subepithelial plasma cells and lymphocyte infiltration; lymphocytes also are present around vessel walls; the predominant infiltrating lymphocyte is the helper T cell
- Immunohistology of the conjunctiva reveals numerous HLA-DR–positive cells in the substantia propria, vessel walls, and epithelium
Management
Most patients with Stevens-Johnson syndrome are treated symptomatically. In principle, the symptomatic treatment of patients with this disorder does not differ from the therapy applied to patients with extensive burns.
Patients should be treated with special attention to airway and hemodynamic stability, fluid status, wound/burn care, and pain control. Therapy for Stevens-Johnson syndrome proceeds as follows:
- Withdrawal of any agent suspected of causing the condition is critically important
- Oral lesions are managed with mouthwashes; topical anesthetics are useful in reducing pain and allowing the patient to take in fluids
- Areas of denuded skin must be covered with compresses of saline or Burow solution
- Tetanus prophylaxis must be addressed
- Extensive debridement of nonviable epidermis followed by immediate cover with biologic dressings is among the recommended treatments.
Ocular therapy
The treatment of acute ocular manifestations usually begins with aggressive lubrication of the ocular surface. As inflammation and cicatricial changes ensue, most ophthalmologists use topical steroids, antibiotics, and symblepharon lysis.
In the case of mild chronic superficial keratopathy, long-term lubrication may be sufficient. In cases of severe ocular involvement, treatment includes the following:
- Removal of keratinized plaques from the posterior lid margins
- Mucous membrane grafting and/or amniotic membrane grafting
- Limbal stem cell transplantation and amniotic membrane grafting
- Superficial keratectomy removing conjunctivalized or keratinized ocular surface
Medication
The goal of pharmacotherapy in patients with Stevens-Johnson syndrome (SJS) is to reduce morbidity and to prevent complications. No specific drug treatment has been consistently shown to be beneficial in the treatment of SJS. The choice of antibiotic for infectious causes depends on the cause of that infection.
Clinical and laboratory evidence suggesting bloodstream infection mandates the use of antibiotics. The most common organisms include Staphylococcus aureus, Pseudomonas aeruginosa, and Enterobacteriaceae species.
The use of systemic corticosteroids is controversial, but may be useful in high doses early in the disease. Morbidity and mortality actually may increase in association with corticosteroid use. For persistent or recurrent ocular inflammation, patients may benefit from short-term systemic corticosteroids and/or long-term immunosuppressive therapy, which may reduce severity of conjunctivitis and improve prognosis quod visum by reducing damage to ocular surface.
Human intravenous immunoglobulin (IVIG) has been described as both treatment and prophylaxis.
Prognosis
Individual lesions typically should heal within 1-2 weeks, unless secondary infection occurs. Most patients recover without sequelae.
Mortality is determined primarily by the extent of skin sloughing. When body surface area (BSA) sloughing is less than 10%, the mortality rate is approximately 1-5%. However, when more than 30% BSA sloughing is present, the mortality rate is between 25% and 35%, and may be as high as 50%. Bacteremia and sepsis appear to play a major role in increased mortality.
The SCORTEN score (a severity-of-illness score for toxic epidermal necrolysis) calculates the risk for death in both SJS and TEN on the basis of the following variables:
- Age >40 years
- Malignancy
- Heart rate >120
- Initial percentage of epidermal detachment >10%
- Blood urea nitrogen (BUN) level >10 mmol/L
- Serum glucose level >14 mmol/L
- Bicarbonate level < 20 mmol/L
Other negative prognostic factors include persistent neutropenia (defined as neutropenia lasting more than 5 days), hypoalbuminemia (usually < 2 g/dL), and persistent azotemia.
In a survival analysis of a cohort of patients with either Stevens-Johnson syndrome or toxic epidermal necrolysis, Sekula et al found that the severity of the cutaneous reaction causing either of these disorders was a risk factor for mortality, but only during the first 90 days following reaction onset.[33] The investigators also found that serious comorbidities and age were risk factors for mortality after 90 days, but not beyond 1 year, past reaction onset. Mortality among patients was 23% at 6 weeks and 34% at 1 year.
Although some patients rapidly progress to lose very large areas of the epidermis in a matter of days, the process suddenly ceases in others and reepithelialization begins a few days later. Predicting the course of disease in a given patient at the initial presentation is not possible. Reepithelialization is usually complete within 3 weeks, but pressure and mucosal areas may remain eroded and crusted for 2 weeks or longer.
Survivors of Stevens-Johnson syndrome may experience numerous long-term sequelae; the most disabling are those of the eye. Cicatrization of conjunctival erosions may lead to the following:
- Inverted eyelashes
- Photophobia
- A burning sensation in the eyes
- Watery eyes
- A siccalike syndrome
- Corneal and conjunctival neovascularization
As many as 40% of survivors of toxic epidermal necrolysis have residual potentially disabling lesions that may cause blindness.